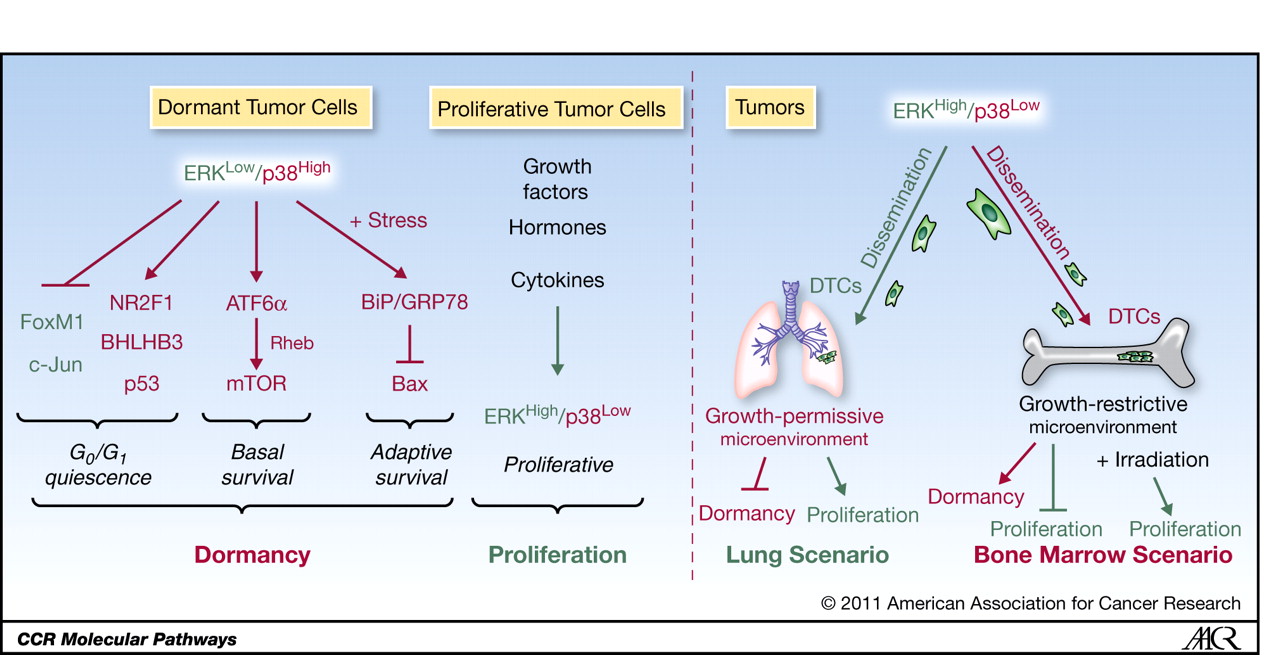
This article by Sosa et al. explores the biology of disseminated tumor cells (DTCs), which are central to cancer relapses and metastasis. DTCs can spread early during tumor development—even from non-invasive lesions—and persist in distant organs such as the bone marrow, liver, and lungs. Many enter a dormant, non-proliferative state, evading therapies that target dividing cells. Clinical dormancy, defined as a prolonged asymptomatic period between treatment and relapse, may be driven by restricted angiogenesis, immune surveillance, or incomplete cellular transformation. Dormancy is actively maintained by metastasis suppressor genes (MSGs), including MKK4, MKK6, Nm23-H1, and BHLHB3, which regulate signaling pathways such as p38 and ERK.
The balance between p38 MAPK and ERK signaling is critical in determining DTC fate. High p38 and low ERK activity promote quiescence through activation of tumor suppressors like p53 and p21, suppression of cyclin D1, and engagement of stress-response programs, including the unfolded protein response (UPR). This signaling profile is shaped by the tumor microenvironment, particularly extracellular matrix (ECM) and integrin signaling. Disruption of integrin–FAK–EGFR pathways leads to p38 activation and deep G0–G1 arrest. Transcription factors downstream of p38, such as BHLHB3 and NR2F1, help sustain this dormant state, allowing DTCs to survive undetected for years.
Recognizing dormancy as an actively maintained state suggests novel therapeutic strategies—not only targeting proliferation but also sustaining dormancy or selectively eliminating dormant DTCs before they reactivate. This study highlights the critical role of the microenvironment and p38/ERK signaling in regulating dormancy and suggests that targeting these pathways may help prevent metastatic relapse.