
by Lucas Unger
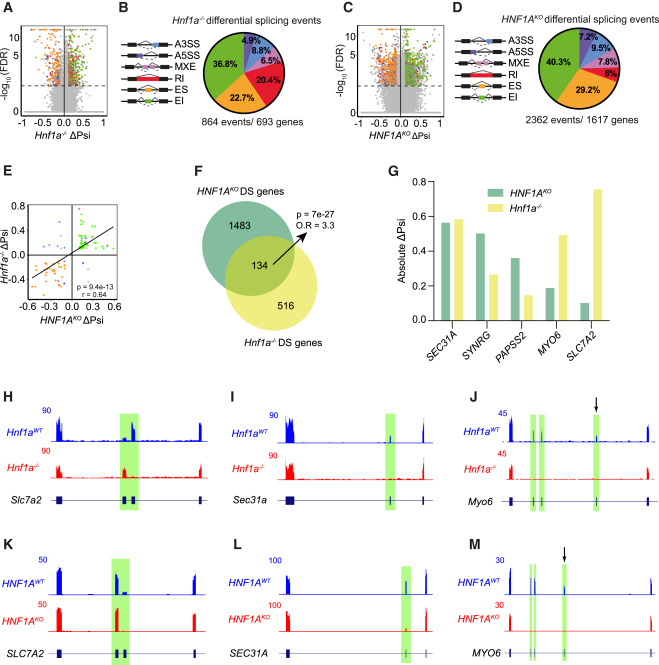
Bernardo et al. demonstrate that HNF1A-deficient diabetes arises primarily from β cell-autonomous defects rather than dysfunction in other HNF1A-expressing tissues. Using tissue-specific Cre recombinase lines with a floxed Hnf1a exon 2 allele, they show that liver-specific (Alb-Cre) and gut-specific (Vil1-Cre) deletions do not cause hyperglycemia, whereas pancreas-specific (Pdx1-Cre) and β cell-specific (Ins1-Cre) deletions recapitulate the diabetic phenotype. The study identifies A1CF as a direct HNF1A target that orchestrates a β cell-specific RNA splicing program affecting genes crucial for insulin secretion, including SLC7A2 which encodes a arginine transporter. This HNF1A-A1CF transcription-splicing axis is suppressed in T2D-associated β cell subpopulations, and genetic variants reducing A1CF expression are linked to increased glycemia and T2D susceptibility. The work establishes a hierarchical regulatory mechanism coordinating β cell transcription and splicing programs essential for glucose homeostasis.
HNF1A regulates AS in mouse and human β cells
Highlights
- HNF1A-deficient diabetes arises from a β cell-autonomous defect
- HNF1A and A1CF coordinate a β cell transcription and splicing program
- Individuals with T2D exhibit increased β cells with suppressed HNF1A–A1CF axis
- Decreased islet A1CF expression is associated with increased glycemia and T2D risk
Continue your reading here:
Bernardo E, De Vas MG, Balboa D, Cuenca-Ardura M, Bonàs-Guarch S, Planas-Fèlix M, Mollandin F, Torrens-Dinarès M, Maestro MA, García-Hurtado J, Moratinos S, Ravassard P, Dou H, Heyn H, van Oudenaarden A, Groen N, de Koning E, Conrad C, Eils R, Vernia S, Rorsman P, Ferrer J. HNF1A and A1CF coordinate a beta cell transcription-splicing axis that is disrupted in type 2 diabetes
Cell Metabolism 2025 Sep 2;37(9):1870-1889.e10. doi: 10.1016/j.cmet.2025.07.007. Epub 2025 Aug 6.PMID: 40774250